- Provides an all-in-one solution for antimicrobial resistance (AMR) data analysis in a One Health approach
- Used in over 175 countries, available in 20 languages
- Generates antibiograms - traditional, combined, syndromic, and even WISCA
- Provides the full microbiological taxonomy and extensive info on all antimicrobial drugs
- Applies all recent CLSI and EUCAST clinical and veterinary breakpoints for MICs, disk zones and ECOFFs
- Corrects for duplicate isolates, calculates and predicts AMR per antimicrobial class
- Integrates with WHONET, ATC, EARS-Net, PubChem, LOINC, SNOMED CT, and NCBI
- 100% free of costs and dependencies, highly suitable for places with limited resources
Now available for Python too! Click here to read more.

Introduction
The AMR package is a free and open-source R package with zero dependencies to simplify the analysis and prediction of Antimicrobial Resistance (AMR) and to work with microbial and antimicrobial data and properties, by using evidence-based methods. Our aim is to provide a standard for clean and reproducible AMR data analysis, that can therefore empower epidemiological analyses to continuously enable surveillance and treatment evaluation in any setting. Many different researchers from around the globe are continually helping us to make this a successful and durable project!
This work was published in the Journal of Statistical Software (Volume 104(3); DOI 10.18637/jss.v104.i03) and formed the basis of two PhD theses (DOI 10.33612/diss.177417131 and DOI 10.33612/diss.192486375).
After installing this package, R knows ~52,000 distinct microbial species (updated December 2022) and all ~600 antimicrobial and antiviral drugs by name and code (including ATC, EARS-Net, ASIARS-Net, PubChem, LOINC and SNOMED CT), and knows all about valid SIR and MIC values. The integral clinical breakpoint guidelines from CLSI and EUCAST are included, even with epidemiological cut-off (ECOFF) values. It supports and can read any data format, including WHONET data. This package works on Windows, macOS and Linux with all versions of R since R-3.0 (April 2013). It was designed to work in any setting, including those with very limited resources. It was created for both routine data analysis and academic research at the Faculty of Medical Sciences of the University of Groningen, in collaboration with non-profit organisations Certe Medical Diagnostics and Advice Foundation and University Medical Center Groningen.
Used in over 175 countries, available in 20 languages
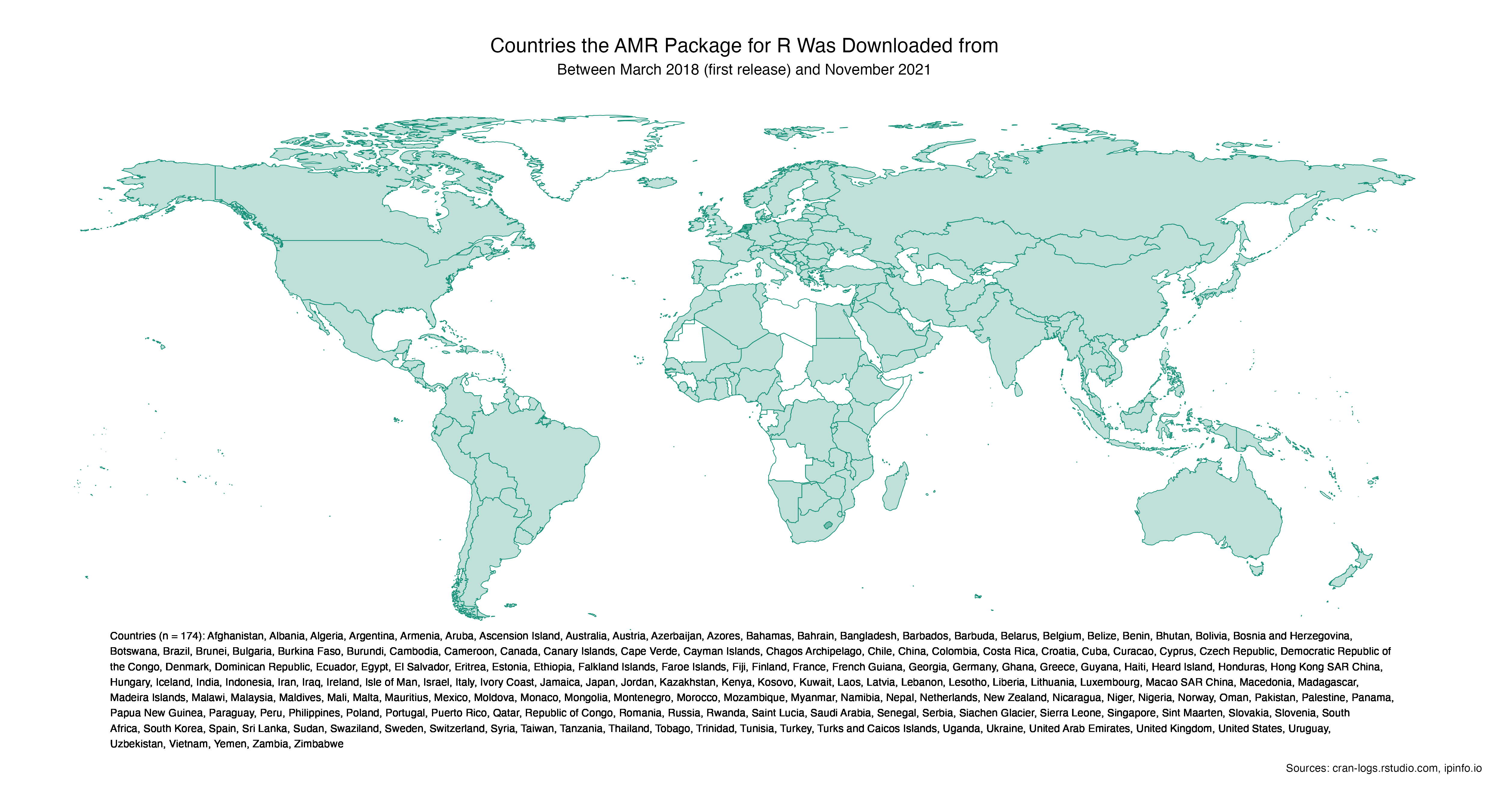
Since its first public release in early 2018, this R package has been used in almost all countries in the world. Click the map to enlarge and to see the country names.
With the help of contributors from all corners of the world, the AMR package is available in  English,
English,  Czech,
Czech,  Chinese,
Chinese,  Danish,
Danish,  Dutch,
Dutch,  Finnish,
Finnish,  French,
French,  German,
German,  Greek,
Greek,  Italian,
Italian,  Japanese,
Japanese,  Norwegian,
Norwegian,  Polish,
Polish,  Portuguese,
Portuguese,  Romanian,
Romanian,  Russian,
Russian,  Spanish,
Spanish,  Swedish,
Swedish,  Turkish, and
Turkish, and  Ukrainian. Antimicrobial drug (group) names and colloquial microorganism names are provided in these languages.
Ukrainian. Antimicrobial drug (group) names and colloquial microorganism names are provided in these languages.
Practical examples
Filtering and selecting data
One of the most powerful functions of this package, aside from calculating and plotting AMR, is selecting and filtering based on antimicrobial columns. This can be done using the so-called antimicrobial selectors, which work in base R, dplyr and data.table.
# AMR works great with dplyr, but it's not required or neccesary
library(AMR)
library(dplyr)
example_isolates %>%
mutate(bacteria = mo_fullname()) %>%
# filtering functions for microorganisms:
filter(mo_is_gram_negative(),
mo_is_intrinsic_resistant(ab = "cefotax")) %>%
# antimicrobial selectors:
select(bacteria,
aminoglycosides(),
carbapenems())With only having defined a row filter on Gram-negative bacteria with intrinsic resistance to cefotaxime (mo_is_gram_negative() and mo_is_intrinsic_resistant()) and a column selection on two antibiotic groups (aminoglycosides() and carbapenems()), the reference data about all microorganisms and all antimicrobials in the AMR package make sure you get what you meant:
| bacteria | GEN | TOB | AMK | KAN | IPM | MEM |
|---|---|---|---|---|---|---|
| Pseudomonas aeruginosa | I | S | R | S | ||
| Pseudomonas aeruginosa | I | S | R | S | ||
| Pseudomonas aeruginosa | I | S | R | S | ||
| Pseudomonas aeruginosa | S | S | S | R | S | |
| Pseudomonas aeruginosa | S | S | S | R | S | S |
| Pseudomonas aeruginosa | S | S | S | R | S | S |
| Stenotrophomonas maltophilia | R | R | R | R | R | R |
| Pseudomonas aeruginosa | S | S | S | R | S | |
| Pseudomonas aeruginosa | S | S | S | R | S | |
| Pseudomonas aeruginosa | S | S | S | R | S | S |
Generating antibiograms
The AMR package supports generating traditional, combined, syndromic, and even weighted-incidence syndromic combination antibiograms (WISCA).
If used inside R Markdown or Quarto, the table will be printed in the right output format automatically (such as markdown, LaTeX, HTML, etc.).
antibiogram(example_isolates,
antimicrobials = c(aminoglycosides(), carbapenems()),
formatting_type = 14)| Pathogen | Amikacin | Gentamicin | Imipenem | Kanamycin | Meropenem | Tobramycin |
|---|---|---|---|---|---|---|
| CoNS | 0% (0-8%) | 86% (82-90%) | 52% (37-67%) | 0% (0-8%) | 52% (37-67%) | 22% (12-35%) |
| E. coli | 100% (98-100%) | 98% (96-99%) | 100% (99-100%) | 100% (99-100%) | 97% (96-99%) | |
| E. faecalis | 0% (0-9%) | 0% (0-9%) | 100% (91-100%) | 0% (0-9%) | 0% (0-9%) | |
| K. pneumoniae | 90% (79-96%) | 100% (93-100%) | 100% (93-100%) | 90% (79-96%) | ||
| P. aeruginosa | 100% (88-100%) | 0% (0-12%) | 100% (88-100%) | |||
| P. mirabilis | 94% (80-99%) | 94% (79-99%) | 94% (80-99%) | |||
| S. aureus | 99% (97-100%) | 98% (92-100%) | ||||
| S. epidermidis | 0% (0-8%) | 79% (71-85%) | 0% (0-8%) | 51% (40-61%) | ||
| S. hominis | 92% (84-97%) | 85% (74-93%) | ||||
| S. pneumoniae | 0% (0-3%) | 0% (0-3%) | 0% (0-3%) | 0% (0-3%) |
In combination antibiograms, it is clear that combined antimicrobials yield higher empiric coverage:
antibiogram(example_isolates,
antimicrobials = c("TZP", "TZP+TOB", "TZP+GEN"),
mo_transform = "gramstain",
formatting_type = 14)| Pathogen | Piperacillin/tazobactam | Piperacillin/tazobactam + Gentamicin | Piperacillin/tazobactam + Tobramycin |
|---|---|---|---|
| Gram-negative | 88% (85-91%) | 99% (97-99%) | 98% (97-99%) |
| Gram-positive | 86% (82-89%) | 98% (96-98%) | 95% (93-97%) |
Like many other functions in this package, antibiogram() comes with support for 20 languages that are often detected automatically based on system language:
antibiogram(example_isolates,
antimicrobials = c("cipro", "tobra", "genta"), # any arbitrary name or code will work
mo_transform = "gramstain",
ab_transform = "name",
formatting_type = 14,
language = "uk") # Ukrainian| Збудник | Гентаміцин | Тобраміцин | Ципрофлоксацин |
|---|---|---|---|
| Грамнегативні | 96% (95-98%) | 96% (94-97%) | 91% (88-93%) |
| Грампозитивні | 63% (60-66%) | 34% (31-38%) | 77% (74-80%) |
Interpreting and plotting MIC and SIR values
The AMR package allows interpretation of MIC and disk diffusion values based on CLSI and EUCAST. Moreover, the ggplot2 package is extended with new scale functions, to allow plotting of log2-distributed MIC values and SIR values.
library(ggplot2)
library(AMR)
# generate some random values
some_mic_values <- random_mic(size = 100)
some_groups <- sample(LETTERS[1:5], 20, replace = TRUE)
interpretation <- as.sir(some_mic_values,
guideline = "EUCAST 2024",
mo = "E. coli", # or any code or name resembling a known species
ab = "Cipro") # or any code or name resembling an antibiotic
# create the plot
ggplot(data.frame(mic = some_mic_values,
group = some_groups,
sir = interpretation),
aes(x = group, y = mic, colour = sir)) +
theme_minimal() +
geom_boxplot(fill = NA, colour = "grey") +
geom_jitter(width = 0.25) +
# NEW scale function: plot MIC values to x, y, colour or fill
scale_y_mic() +
# NEW scale function: write out S/I/R in any of the 20 supported languages
# and set colourblind-friendly colours
scale_colour_sir() 
Calculating resistance per group
For a manual approach, you can use the resistance or susceptibility() function:
example_isolates %>%
# group by ward:
group_by(ward) %>%
# calculate AMR using resistance() for gentamicin and tobramycin
# and get their 95% confidence intervals using sir_confidence_interval():
summarise(across(c(GEN, TOB),
list(total_R = resistance,
conf_int = function(x) sir_confidence_interval(x, collapse = "-"))))| ward | GEN_total_R | GEN_conf_int | TOB_total_R | TOB_conf_int |
|---|---|---|---|---|
| Clinical | 0.2289362 | 0.205-0.254 | 0.3147503 | 0.284-0.347 |
| ICU | 0.2902655 | 0.253-0.33 | 0.4004739 | 0.353-0.449 |
| Outpatient | 0.2000000 | 0.131-0.285 | 0.3676471 | 0.254-0.493 |
Or use antimicrobial selectors to select a series of antibiotic columns:
library(AMR)
library(dplyr)
out <- example_isolates %>%
# group by ward:
group_by(ward) %>%
# calculate AMR using resistance(), over all aminoglycosides and polymyxins:
summarise(across(c(aminoglycosides(), polymyxins()),
resistance))
out| ward | GEN | TOB | AMK | KAN | COL |
|---|---|---|---|---|---|
| Clinical | 0.229 | 0.315 | 0.626 | 1 | 0.780 |
| ICU | 0.290 | 0.400 | 0.662 | 1 | 0.857 |
| Outpatient | 0.200 | 0.368 | 0.605 | 0.889 |
# transform the antibiotic columns to names:
out %>% set_ab_names()| ward | gentamicin | tobramycin | amikacin | kanamycin | colistin |
|---|---|---|---|---|---|
| Clinical | 0.229 | 0.315 | 0.626 | 1 | 0.780 |
| ICU | 0.290 | 0.400 | 0.662 | 1 | 0.857 |
| Outpatient | 0.200 | 0.368 | 0.605 | 0.889 |
# transform the antibiotic column to ATC codes:
out %>% set_ab_names(property = "atc")| ward | J01GB03 | J01GB01 | J01GB06 | J01GB04 | J01XB01 |
|---|---|---|---|---|---|
| Clinical | 0.229 | 0.315 | 0.626 | 1 | 0.780 |
| ICU | 0.290 | 0.400 | 0.662 | 1 | 0.857 |
| Outpatient | 0.200 | 0.368 | 0.605 | 0.889 |
What else can you do with this package?
This package was intended as a comprehensive toolbox for integrated AMR data analysis. This package can be used for:
- Reference for the taxonomy of microorganisms, since the package contains all microbial (sub)species from the List of Prokaryotic names with Standing in Nomenclature (LPSN) and the Global Biodiversity Information Facility (GBIF) (manual)
- Interpreting raw MIC and disk diffusion values, based on any CLSI or EUCAST guideline (manual)
- Retrieving antimicrobial drug names, doses and forms of administration from clinical health care records (manual)
- Determining first isolates to be used for AMR data analysis (manual)
- Calculating antimicrobial resistance (tutorial)
- Determining multi-drug resistance (MDR) / multi-drug resistant organisms (MDRO) (tutorial)
- Calculating (empirical) susceptibility of both mono therapy and combination therapies (tutorial)
- Predicting future antimicrobial resistance using regression models (tutorial)
- Getting properties for any microorganism (like Gram stain, species, genus or family) (manual)
- Getting properties for any antimicrobial (like name, code of EARS-Net/ATC/LOINC/PubChem, defined daily dose or trade name) (manual)
- Plotting antimicrobial resistance (tutorial)
- Applying EUCAST expert rules (manual)
- Getting SNOMED codes of a microorganism, or getting properties of a microorganism based on a SNOMED code (manual)
- Getting LOINC codes of an antibiotic, or getting properties of an antibiotic based on a LOINC code (manual)
- Machine reading the EUCAST and CLSI guidelines from 2011-2021 to translate MIC values and disk diffusion diameters to SIR (link)
- Principal component analysis for AMR (tutorial)
Get this package
Latest official version
This package is available here on the official R network (CRAN). Install this package in R from CRAN by using the command:
install.packages("AMR")It will be downloaded and installed automatically. For RStudio, click on the menu Tools > Install Packages… and then type in “AMR” and press Install.
Note: Not all functions on this website may be available in this latest release. To use all functions and data sets mentioned on this website, install the latest development version.
Latest development version
![]()
![]()
![]()
Please read our Developer Guideline here.
The latest and unpublished development version can be installed from the rOpenSci R-universe platform:
install.packages("AMR", repos = "https://msberends.r-universe.dev")Get started
To find out how to conduct AMR data analysis, please continue reading here to get started or click a link in the ‘How to’ menu.
Partners
The development of this package is part of, related to, or made possible by the following non-profit organisations and initiatives:
![]()
![]()
![]()
![]()
![]()
Copyright
This R package is free, open-source software and licensed under the GNU General Public License v2.0 (GPL-2). In a nutshell, this means that this package:
May be used for commercial purposes
May be used for private purposes
May not be used for patent purposes
-
May be modified, although:
- Modifications must be released under the same license when distributing the package
- Changes made to the code must be documented
-
May be distributed, although:
- Source code must be made available when the package is distributed
- A copy of the license and copyright notice must be included with the package.
Comes with a LIMITATION of liability
Comes with NO warranty